Билет 12
3. Спектроскопические методы диагностики наноструктур (инфракрасная спектроскопия, рамановская спектроскопия, флуоресцентная и люминесцентная, фотоэмиссионная магнито-резонансная (ЭПР, ЯМР)).
Инфракрасная спектроскопия
Этот метод анализа основан на записи инфракрасных спектров поглощения вещества. Поглощение веществом в области инфракрасного излучения происходят за счёт так колебаний атомов в молекулах. Колебания подразделяются на валентные (когда в ходе колебания изменяются расстояния между атомами) и колебательные (когда в ходе колебания изменяются углы между связями).
Переходы между различными колебательными состояниями в молекулах квантованы, благодаря чему поглощение в ИК-области имеет форму спектра, где каждому колебанию соответствует своя длина волны. Понятно что длина волны для каждого колебания зависит от того какие атомы в нём участвуют, и кроме того она мало зависит от их окружения. То есть для каждой функциональной группы (С=О, О-Н, СН2 и пр) характерны колебания определённой длины волны, точнее говоря даже для каждой группы характерен ряд колебаний (соответственно и полос в ИК-спектре). Именно на этих свойствах ИК-спектров основана идентификация соединений по спектральным данным. Однако не всё так просто. Во первых метод ИК-спектроскопии не являете разделяющим методом, то есть при исследовании какого-либо вещества может оказаться что исследовалась на самом деле смесь нескольких веществ, что конечно сильно исказит результаты расшифровки спектра. Ну и всё ж говорить об однозначной идентификации вещества с помощью метода ИК-спектроскопии не вполне правильно, так как метод скорее позволяет выявить определённые функциональные группы, а не их количество в соединении и их способ связи друг с другом.
ЛЮМИНЕСЦЕНЦИЯ
(от лат. lumen, род. падеж luminis -свет и -escent - суффикс, означающий слабое действие), свечение в-ва, возникающее после поглощения им энергии возбуждения.
Представляет собой избыток над тепловым излучением, испускаемым в-вом при данной т-ре за счет его внутренней (тепловой) энергии. В отличие от др. видов свечения (напр., рассеяния света, тормозного излучения) Л. характеризуется временем свечения, значительно превышающим период колебаний световой волны и составляющим от 10-12 с до неск. суток.
Понятие Л. применимо только к такому в-ву (совокупности частиц), состояние к-рого не слишком отличается от термодинамически равновесного, иначе различие между Л. и тепловым излучением теряет смысл. Механизм Л. заключается в образовании под действием энергии от внеш. или внутр. источника возбужденных состояний атомов, молекул, кристаллов и послед. испускании ими квантов света (фотонов).
По типу возбуждения выделяют фотолюминесценцию (источник энергии возбуждения - свет), радиолюминесценцию (радиоактивное излучение), рентгенолюминесценцию (рентгеновское излучение), электролюминесценцию (электрич. поле), катодолюминесценцию (пучок электронов), триболюминесценцию (мех. воздействие), хемилюминесценцию (хим. р-ции) и др. Различают молекулярную Л., при к-рой молекулы или атомы испускают фотоны при переходе из возбужденного состояния в основное квантовое состояние, и рекомбинационную Л., когда под действием энергии возбуждения образуются носители заряда (электроны и дырки в кристаллофосфорах) или ионы и радикалы (в газах, жидкостях, стеклах), послед. рекомбинация к-рых сопровождается испусканием фотонов. Излучат. переход из возбужденного состояния в основное происходит самопроизвольно (спонтанная Л.) или под действием внеш. электромагн. излучения (вынужденная Л.).
Испускание света может происходить не обязательно теми же молекулами, к-рые возбуждаются при поглощении энергии, но и другими, если происходит безызлучат. передача энергии возбуждения (сенсибилизированная Л.).
Л. характеризуют спектром испускания (фотолюминесценцию - также спектром возбуждения), квантовым выходом, поляризацией, кинетикой затухания. В данной статье рассматривается мол. фотолюминесценция, к-рую широко применяют в технике и аналит. химии (см. Люминофоры, Люминесцентный анализ), фотохимии и хим. кинетике для изучения св-в возбужденных состояний частиц и очень быстрых хим. р-ций, в фотобиологии, биохимии и медицине для изучения св-в биол. объектов и механизма биол. процессов.
Механизм Л.
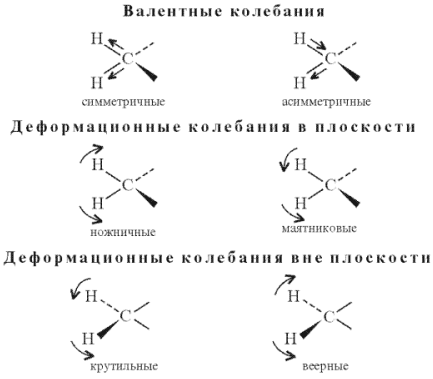
Молекулярную фотолюминесценцию подразделяют на флуоресценцию и фосфоресценцию. Флуоресценция характеризуется малой длительностью (менее 10-6 с) и обусловлена испусканием фотонов при переходе системы из возбужденного состояния той же мультиплетности, что и основное состояние. Фосфоресценция -длит. свечение (от долей до неск. десятков с), к-рое возникает при переходе в осн. состояние из возбужденного состояния иной мультиплетности; такой переход происходит с нарушением спинового правила отбора (см. Квантовые переходы). Для большинства орг. молекул с четным числом электронов осн. состояние является синглетным, а низшие возбужденные состояния имеют мультиплетность 1 и 3, т. е. могут быть синглетными и триплетными. Для таких молекул флуоресценция представляет собой излучат. переход в осн. состояние S0 из возбужденного синглетного состояния S1 (переход 2 на рис. 1).

Рис. 1. Схема квантовых переходов при молекулярной люминесценции. S0 - основной электронный уровень (с колебат. уровнями), S1 и Т1 - возбужденные электронные уровни (синглетный и триплетный соотв.). Прямыми вертикальными стрелками обозначены: поглощение (1), излучательные переходы флуоресценция (2) и фосфоресценция (3), горизонтальными стрелками - безызлучат. переходы: интеркомбинац. конверсия (4) и внутр. конверсия (5). Волнистыми стрелками обозначены процессы колебат. релаксации энергии возбуждения.
С флуоресценцией конкурирует безызлучат. переход в триплетное состояния Т1 с энергией меньшей, чем у состояния S1 (интеркомбинац. конверсия). Фосфоресценция - излучат. переход из ниж. триплетного состояния Т1 в осн. состояние - наблюдается в условиях, когда конкурирующие с данным излучат. переходом безызлучат. процессы замедлены (высокая вязкость в-ва, низкие т-ры и т. п.).
Возможна и т.наз. замедленная флуоресценция, когда вследствие, напр., термич. активации молекул в возбужденном триплетном состоянии T1 происходит безызлучат. переход в возбужденное синглетное состояние S1 с послед. испусканием фотона в результате излучат. перехода S1 : S0. Спектр замедленной флуоресценции идентичен спектру обычной флуоресценции, но время затухания на неск. порядков больше. У нек-рых молекул осн. состояние не является синглетным. Так, для О2 осн. состояние триплетное 3Sg слабая фосфоресценция, наблюдаемая для этого в-ва в ближней ИК области, обусловлена переходом из ниж. синглетного состояния 1D в основное. Для радикалов, имеющих один неспаренный электрон, осн. состояние дублетное (мультиплетность 2), низшие возбужденные состояния имеют мультиплетность 2 и 4 (соотв. дублетные и квартетные состояния). Флуоресценция радикалов наблюдается при переходе из ниж. возбужденного дублетного состояния в основное.
 Физическая
природа люминесценции состоит в излучательных переходах атомов или молекул (электронов) из возбуждённого состояния в основное.
Физическая
природа люминесценции состоит в излучательных переходах атомов или молекул (электронов) из возбуждённого состояния в основное.
Рис.2. Схема квантовых переходов в элементарном акте поглощение - люминесценция, 1 — основной энергетический уровень; 2 — уровень излучения; 3 — уровень возбуждения, переход 1 — 3 поглощение кванта, переход 3 — 1 резонансная люминесценция, переход 2 — 1 спонтанная люминесценция, переход 3 — 2 безизлучательный переход.
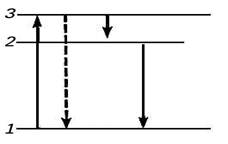
В кристаллофосфорах возбуждение светом, электрическим током или пучком частиц создаёт свободные электроны, дырки и экситоны (рис. 3). Электроны могут мигрировать по решётке, оседая на ловушках 4. Л., происходящая при рекомбинации свободных электронов с дырками, называется межзонной (а). Если рекомбинирует электрон с дыркой, захваченной центром свечения (атомом примеси или дефектом решётки), происходит Л. центра (б). Рекомбинация экситонов даёт экситонную люминесценцию. (в). Спектр люминесценции. кристаллофосфоров состоит из межзонной, экситонной и примесной полос.
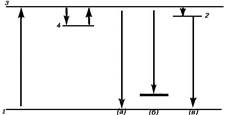 Рис. 3. Схема энергетических переходов при
люминесценции кристаллофосфоров: 1 — валентная зона, 3 — зона проводимости.
Переход 1—3 соответствует поглощению энергии, переходы 3—4 и 4—3 — захвату и
освобождению электрона метастабильным уровнем (ловушкой 4). Переход (а)
соответствует межзонной люминесценции, (б) — люминесценции центра, (в) —
экситонной люминесценции (2 — уровень энергии экситона).
Рис. 3. Схема энергетических переходов при
люминесценции кристаллофосфоров: 1 — валентная зона, 3 — зона проводимости.
Переход 1—3 соответствует поглощению энергии, переходы 3—4 и 4—3 — захвату и
освобождению электрона метастабильным уровнем (ловушкой 4). Переход (а)
соответствует межзонной люминесценции, (б) — люминесценции центра, (в) —
экситонной люминесценции (2 — уровень энергии экситона).
ЭЛЕКТРОННЫЙ ПАРАМАГНИТНЫЙ РЕЗОНАНС (ЭПР, электронный спиновый резонанс), явление резонансного поглощения электромагн. излучения парамагн. частицами, помещенными в постоянное магн. поле; один из методов радиоспектроскопии. Используется для изучения систем с ненулевым электронным спиновым магн. моментом (т. е. обладающих одним или неск. неспаренными электронами): атомов, своб. радикалов в газовой, жидкой и твердой фазах, точечных дефектов в твердых телах, систем в триплетном состоянии, ионов переходных металлов.
Физика
явления. В
отсутствие постоянного магн. поля Н магн. моменты неспаренных электронов направлены
произвольно, состояние системы таких частиц вырождено по энергии. При наложении
поля Н проекции магн. моментов на направление поля принимают определенные
значения и вырождение снимается (см. Зеемана эффект), т. е. происходит расщепление
уровня энергии электронов E0.
Расстояние между возникшими подуровнями зависит от напряженности поля Н и равно![]() (рис. 1),
где g - фактор спектроскопич. расщепления (см. ниже),
(рис. 1),
где g - фактор спектроскопич. расщепления (см. ниже),![]() -
магнетон Бора, равный
9,274 x 10-24 Дж/Тл; в
системе единиц СИ вместо Н следует использовать магн. индукцию
-
магнетон Бора, равный
9,274 x 10-24 Дж/Тл; в
системе единиц СИ вместо Н следует использовать магн. индукцию![]() где
где![]() - магн.
проницаемость своб. пространства, равная 1,257 x 10-6 Гн/м. Распределение электронов по
подуровням подчиняется закону Больцмана, согласно к-рому отношение
заселенностей подуровней определяется выражением
- магн.
проницаемость своб. пространства, равная 1,257 x 10-6 Гн/м. Распределение электронов по
подуровням подчиняется закону Больцмана, согласно к-рому отношение
заселенностей подуровней определяется выражением![]() где k - постоянная
Больцмана, Т - абс. т-ра. Если на образец подействовать переменным магн.
полем с частотой v, такой, что
где k - постоянная
Больцмана, Т - абс. т-ра. Если на образец подействовать переменным магн.
полем с частотой v, такой, что![]() (h - постоянная Планка), и направленным перпендикулярно
H, то индуцируются переходы между соседними подуровнями, причем переходы с
поглощением и испусканием кванта hv равновероятны. Т.к. на нижнем уровне числоэлектронов больше в
соответствии с распределением Больцмана, то преим. будет происходить
резонансное поглощение энергии переменного магн. поля (его магн. составляющей).
(h - постоянная Планка), и направленным перпендикулярно
H, то индуцируются переходы между соседними подуровнями, причем переходы с
поглощением и испусканием кванта hv равновероятны. Т.к. на нижнем уровне числоэлектронов больше в
соответствии с распределением Больцмана, то преим. будет происходить
резонансное поглощение энергии переменного магн. поля (его магн. составляющей).
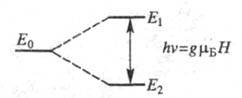
Рис. 1. Расщепление энергетического уровня электрона в постоянном магнитном поле. Е0 - уровень в отсутствие поля, Е1 и Е2 - уровни, возникающие в присутствии поля Н.
Для непрерывного наблюдения поглощения энергии условия резонанса недостаточно, т.к. при воздействии электро-магн. излучения произойдет выравнивание заселенностей подуровней (эффект насыщения). Для поддержания больцманов-ского распределения заселенностей подуровней необходимы релаксационные процессы. Релаксационные переходы электронов из возбужденного состояния в основное реализуются при обмене энергией с окружающей средой (решеткой), к-рый осуществляется при индуцированных решеткой переходах между электронными подуровнями и определяется как спин-решеточная релаксация. Избыток энергии перераспределяется и между самими электронами - происходит спин-спиновая релаксация. Времена спин-решеточной релаксации T1 и спин-спиновойрелаксации Т2 являются количеств. мерой скорости возврата спиновой системы в исходное состояние после воздействия электромагн. излучения. Зафиксированное регистрирующим устройством поглощение электромагн. энергии спиновой системой и представляет собой спектр ЭПР.
Техника эксперимента. В спектроскопии ЭПР используют радиоспектрометры, принципиальная блок-схема к-рых представлена на рис. 6. В серийных приборах частота электромагн. излучения задается постоянной, а условие резонанса достигается путем изменения напряженности магн. поля. Большинство спектрометров работает на частоте v 9000 МГц, длина волны 3,2 см, магн. индукция 0,3 Тл. Электромагн. излучение сверхвысокой частоты (СВЧ) от источника К по волноводам В поступает в объемный резонатор Р, содержащий исследуемый образец и помещенный между полюсами электромагнита NS.
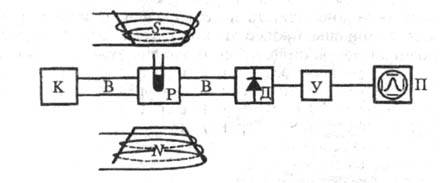
Рис. 6. Блок-схема спектрометра ЭПР. К - источник СВЧ излучения, В -волноводы, Р - объемный резонатор, Д - детектор СВЧ излучения, У - усилитель, NS - электромагнит, П - регистрирующее устройство.
В
условиях резонанса СВЧ излучение поглощается спиновой системой. Модулированное
поглощением СВЧ излучение по волноводу (В) поступает на детектор Д. После
детектирования сигнал усиливается на усилителе У и подается на регистрирующее
устройство П. В этих условиях регистрируется и интегральная линия поглощения
ЭПР. Для повышения чувствительности и разрешения спектрометров ЭПР используют
высокочастотную (ВЧ) модуляцию (обычно 100 кГц) внешнего магн. поля, осуществляемую
с помощью модуляционных катушек. ВЧ модуляция и спец. фазочувст-вит.
детектирование преобразуют сигнал ЭПР в первую производную кривой поглощения, в
виде к-рой и происходит регистрация спектров ЭПР в большинстве серийных
спектрометров. В нек-рых спец. случаях используют спектрометры, работающие в
диапазоне длин волн 8 мм и 2 мм, что позволяет существенно улучшить разрешение
по g-фактору (своб. радикалы, парамагн. ионы).
Чувствительность совр. спектрометров достигает 10-9 М (1011 частиц в образце) при оптимальных условиях
регистрации и ширине линии 10-4 Тл. Важной характеристикой является временная
шкала метода, определяемая частотой СВЧ излучения, подающегося на образец (v =
10-10 с), что
позволяет исследовать динамику в спиновых системах в диапазоне частот 106-1010 c-1.
Применение. Методом ЭПР можно определять концентрацию и идентифицировать
парамагн. частицы в любом агрегатном состоянии, что
незаменимо для исследования кинетики и механизма процессов, происходящих с их
участием. Спектроскопия ЭПР
применяется врадиационной химии, фотохимии, катализе, в изучении процессов окисления и горения, строения и реакционной способности орг. своб. радикалов и ион-радикалов, полимерных систем с
сопряженными связями. Методом ЭПР решается широкий круг структурно-динамич.
задач. Детальное исследование спектров ЭПР парамагн. ионов d- и
f-элементов позволяет определить валентное состояние иона, найтисимметрию кристаллич.
поля, количественно изучать кинетику и термодинамику многоступенчатых
процессов комплексообразованияионов. Динамич. эффекты в спектрах ЭПР, проявляющиеся в специфич.
уширении отдельных компонент СТС, обусловленном модуляцией величины констант СТВ за
счет внутри- и межмол. хим. р-ций, позволяют количественно исследовать эти
р-ции, напр. электронный обмен между ион-радикалами и
исходными молекулами типа А-*+
А![]() А + А-*,
лигандный обмен типа LR* + L'
А + А-*,
лигандный обмен типа LR* + L'![]() L'R*+L, внутримол. процессы вращения
отдельных фрагментов в радикалах, конформац. вырожденные переходы, внутримол.
процессы перемещения атомовили групп атомов в радикалах и т.д.
L'R*+L, внутримол. процессы вращения
отдельных фрагментов в радикалах, конформац. вырожденные переходы, внутримол.
процессы перемещения атомовили групп атомов в радикалах и т.д.
КОМБИНАЦИОННОГО РАССЕЯНИЯ СПЕКТРОСКОПИЯ (рамановская спектроскопия), раздел оптич. спектроскопии,
изучающий взаимод. монохроматич. излучения с в-вом, сопровождающееся изменением
энергии рассеянного излучения по сравнению с энергией падающего на объект
(возбуждающего) излучения. Комбинац. рассеяние (КР) обусловлено неупругими
столкновениями фотонов с молекулами (илиионами), в ходе к-рых
они обмениваются энергией. По изменению энергии фотона можно судить об
изменении энергии молекулы, т.е. о
переходе ее на новый энергетич. уровень. Схематически эти переходы показаны на
рис. 1. Молекула, находящаяся в
невозбужденном состоянии с
энергией Е0, под действием кванта с энергией hv0 (h-постоянная Планка, v0-частота
падающего кванта) возбуждается в промежуточное (виртуальное) состояние с
энергией Eвиpт, откуда может либо вернуться в исходное состояние,
испустив квант hv0(рэлеевское рассеяние), либо перейти в состояние Еi,
испустив квант h(v0—vi), что приводит к появлению в
спектре рассеянного излучения линий с частотами v0—vi (стоксовы линии). Если до поглощения
фотона молекула находилась в возбужденном состоянии с энергией Ei,
то после рассеяния света она может перейти как в исходное, так и в основное
состояние E0, тогда энергия
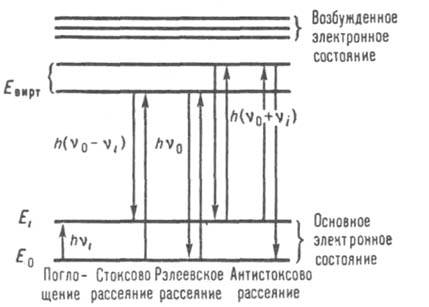
Рис. 1. Схема энергетических уровней,
иллюстрирующая основные принципы КР. Энергия возбуждающего света hv0,
линии КР имеют частоты v0bvi.
рассеянного
света возрастает, составляя h(v0+vi), что приводит к
появлению в спектре линий с частотами v0+vi (антистоксовы линии). Энергетич.
состояние в-ва характеризует разность энергий возбуждающего и рассеянного света
Ei+hvi, т. е, важнейшей характеристикой спектров КР
являются не сами частоты, а их сдвиг относительно частоты рэлеевской линии.
Стоксовы и антистоксовы линии располагаются симметрично относительно рэлеевской
линии и образуют спектр КР; при этом сдвиги частот vi имеют значения 10-4000 см-1 и совпадают с частотами молекул, наблюдаемыми в ИК спектрах поглощения. Спектр КР, как правило, представляет
собой колебат. спектр. В области малых значений vi могут проявляться переходы между
вращат. уровнями (вращат. спектры КР), реже электронные переходы (электронные спектры КР). Т.
обр., частоты рассеянного света являются комбинациями частоты возбуждающего
света и колебат. и вращат. частотмолекул. При обычной т-ре стоксовы линии значительно
интенсивнее антистоксовых, поскольку б.ч. молекул находится
в невозбужденном состоянии; при повышении т-ры интенсивность антистоксовых
линий растет из-за частичного теплового заселения возбужденных колебат.
состояний Ei. Интенсивность стоксовых линий КР пропорциональна (vo-vi)4 при vo<<vэл (vэл - частота электронного перехода), а
при v0:vэлрезко возрастает (резонансное КР). Для каждой
конкретной линии КР интенсивность - ф-ция поляризуемости молекул (a), в
отличие от ИК поглощения, где интенсивность - ф-ция дипольного момента молекулы (m).
Значение наведенного дипольного момента определяется
выражением
![]()
где Е - напряженность электрич. поля световой волны, a, b, g поляризуемость I, II,
III порядка. В случае обычного КР вторым и остальными членами разложения можно
пренебречь, однако при больших интенсивностях возбуждающего света они имеют
важное значение. Различия в физ. природе процессов рассеяния и поглощения света
характеризуют разные правила отбора, т. е. одни и те же колебания
проявляются либо в ИК, либо в КР спектрах или имеют разную интенсивность,
напр., для молекул, обладающих центром симметрии,
активные в спектре КР колебания не проявляются в ИК спектре и наоборот (правило
альтернативного запрета); колебат. и вращат. частоты простых бездипольных молекул (Н2,
О2, N2 и
др.), активные в спектрах КР, в ИК спектрах отсутствуют. Таким образом спектры
ИК и КР дополняют друг друга. При
КР происходит изменение поляризации света,
характеризуемое степенью деполяризации r. При
использовании для возбуждения лазера (рис. 2),
излучение к-рого поляризовано в плоскости ху, r=Iz/Ix,
где Iz и Ix - интенсивности компонент
рассеянного света, поляризованных в направлении осей z и х соответственно. Для
неполносимметричных колебаний (хаотически ориентир, молекул в газе или жидкой фазе) r=0,75 (деполяризов. линии в
спектре); для
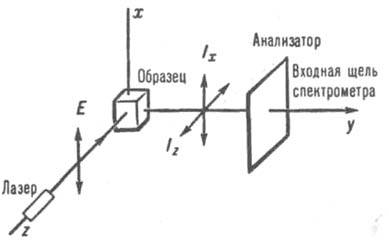
Рис. 2. Наблюдение спектра КР под углом 90° при возбуждении поляризованным
светом.
полносимметричных колебаний 0[r[0,75 (поляризов. линии), что позволяет использовать величину r для установления симметрии молекул. При возбуждении неполяризов. светом для неполносимметричных колебаний r=0,86. . Комбинационного рассеяния спектроскопияприменяется для изучения орг. и неорг. в-в в любых агрегатных состояниях, за исключением черных и глубоко окрашенных образцов и соед., обладающих сильной флуоресценцией в видимой области спектра. По сравнению с И К спектрами имеет преимущества при исследовании водных р-ров, тонких волокон, микрообъектов, при изучении низкочастотных колебаний. Комбинационного рассеяния спектроскопиюиспользуют для идентификации в-в, определения отдельных хим. связей и групп в молекулах, для исследования внутри- и межмол. взаимодействий, разл. видов изомерии, фазовых переходов, водородных связей, адсорбир. молекул и катализаторов, для обнаружения микропримесей в-в, загрязняющих окружающую среду. Использование лазеров значительно расширило границы применения комбинационного рассеянии спектроскопия и привело к развитию ряда новых методов в спектроскопии КР. Возможность изменения длины волны возбуждения путем замены лазеров или с помощью лазера с перестраиваемой частотой привела к развитию резонансного КР, к-рое возникает, когда частота возбуждающего света попадает в область поглощения в-ва. Этот метод позволяет определять низкиеконцентрации в-в, что особенно важно для биологии и биохимии. При возбуждении КР лазерами большой мощности наблюдаются новые эффекты, обусловленные нелинейными членами в разложении (1). Гипер - КР связан с гиперполяризуемостью b, наблюдается в области частот 2(vobvi) и позволяет измерять частоты колебаний, запрещенных и в КР, и в ИК спектрах; кроме того, в гипер - КР проявляются все ИК активные колебания, к-рые м. б. легко идентифицированы, т.к. они поляризованы. Когерентное антистоксово рассеяние света (КАРС) связано с третьим членом в разложении (1), содержащим поляризуемость третьего порядка g. При одновременном облучении образца двумя лазерами с частотами v1 и v2, направленными под небольшим углом, и если разность v1-v2=vi совпадает с одной из внутримол. частот, на частоте 2(v1-v2) возникает направленное лазероподобное излучение, интенсивность к-рого значительно выше интенсивности обычного КР. Плавно меняя частоту v2, можно получить весь спектр КАРС. Этот метод м. 6. использован для анализа в-в при высокой т-ре.Под действием мощных лазеров может возникнуть также вынужденное КР, при к-ром рассеянные фотоны стимулируют (вынуждают) дальнейший процесс рассеяния. Интенсивность отдельных линий при этом резко возрастает и делается сравнимой с интенсивностью возбуждающего света. При одновременном облучении образца интенсивным лазерным пучком с частотой v0 и непрерывным белым излучением с частотами в интервале от v0 до v0+4000 см-1 возникает спектр инверсного КР. При этом в спектрах поглощения наблюдаются частоты активные в КР. Новые возможности для исследования структуры оптически активных молекул в области колебат. переходов открывает спектр кругового дихроизма КР, представляющий собой разность спектров, полученных при возбуждении КР излучением, поляризованным по кругу вправо и влево. Обнаружение резкого усиления (до 106 раз) интенсивности КР молекул на пов-сти нек-рыхметаллов (Ag, Au, Сu), т. наз. гигантское КР, позволяет исследовать процессы адсорбции и гетерог. катализа. В настоящее время выпускают спектрометры, к-рые регистрируют спектры КР бесцв. и окрашенных образцов в кол-вах до 10-4 г (или мл). Разработаны скоростные спектрометры с использованием импульсных лазеров, регистрирующие спектр КР за 10-9 с, а также приборы, к-рые сочетаютлазер с микроскопом и позволяют получать спектры КР от объектов размером порядка 1 мкм. КР открыт в 1928 Л. И. Мандельштамом и Г. С. Ландсбергом (СССР) для кристаллов и независимо от них Ч. В. Раманом и К. С. Кришнаном (Индия) для жидкостей.
Я́дерный магни́тный резона́нс (ЯМР) — резонансное поглощение или излучение электромагнитной энергии веществом, содержащим ядра с ненулевым спином во внешнем магнитном поле, на частоте ν (называемой частотой ЯМР), обусловленное переориентацией магнитных моментов ядер.
Одни и те же ядра атомов в различных окружениях в молекуле показывают различные сигналы ЯМР. Отличие такого сигнала ЯМР от сигнала стандартного вещества позволяет определить так называемый химический сдвиг, который обусловлен химическим строением изучаемого вещества. В методиках ЯМР есть много возможностей определять химическое строение веществ, конформации молекул, эффекты взаимного влияния, внутримолекулярные превращения.
Спектроскопи́я я́дерного магни́тного резона́нса, ЯМР-спектроскопия — спектроскопический метод исследования химических объектов, использующий явление ядерного магнитного резонанса. Наиболее важными для химии и практических применений являются спектроскопия протонного магнитного резонанса (ПМР-спектроскопия), а также спектроскопия ЯМР на ядрах углерода-13 (13C ЯМР-спектроскопия), фтора-19 (19F ЯМР-спектроскопия), фосфора-31 (31P ЯМР-спектроскопия).
Подобно инфракрасной спектроскопии, ЯМР выявляет информацию о молекулярном строении химических веществ. Однако, он обеспечивает более полную информацию, чем ИС, позволяя изучать динамические процессы в образце — определять константы скорости химических реакций, величину энергетических барьеров внутримолекулярного вращения. Эти особенности делают ЯМР-спектроскопию удобным средством как в теоретической органической химии, так и для анализа биологических объектов.
Базовая ЯМР техника
ЯМР образец помещается в тонкостенную стеклянную трубку. Когда ее помещают в магнитное поле, ЯМР активные ядра (такие как 1H или 13C) поглощают электромагнитную энергию. Резонансная частота, энергия абсорбции и интенсивность испущенного сигнала пропорциональны силе магнитного поля. Так в поле в 21 Тесла, протон резонирует при частоте 900 МГц.
[править]Химический сдвиг
Основная статья: Химический сдвиг
В зависимости от местного электронного окружения, разные протоны в молекуле резонируют на слегка отличающихся частотах. Так как и это смещение частоты и основная резонансная частота прямо пропорциональны силе магнитного поля, то это смещение преобразуется в независимую от магнитного поля безразмерную величину известную как химический сдвиг. Химический сдвиг определяется как относительное изменение относительно некоторых эталонных образцов. Частотный сдвиг экстремально мал в сравнении с основной ЯМР частотой. Типичный сдвиг частоты равен 100 Гц, тогда как базовая ЯМР частота имеет порядок 100 МГц. Таким образом химический сдвиг часто выражается в частях на миллион (ppm). Для того что обнаружить такое маленькое различие частоты, приложенное магнитное поле должно быть постоянным внутри объема образца.
Так как химический сдвиг зависит от химического строения вещества, он применяется для получения структурной информации о молекулах в образце. К примеру, спектр дляэтанола (CH3CH2OH) дает 3 отличительных сигнала, то есть 3 химических сдвига: один для группы CH3, второй для СН2-группы и последний для OH. Типичный сдвиг для CH3-группы примерно равен 1 ppm, для CH2-группы присоединенной к OH-4 ppm и OH примерно 2—3 ppm.
Из-за молекулярного движения при комнатной температуре 3 метиловых протона вылетают в среднем в течение ЯМР процесса, который длится лишь несколько миллисекунд. Эти протоны вырождаются и формируют пики при том же химическом сдвиге. Программное обеспечение позволяет проанализировать размер пиков для того, чтобы понять как много протонов дает вклад в эти пики.
[править]Спин-спиновое взаимодействие
Наиболее полезную информацию для определения структуры в одномерном ЯМР-спектре даёт так называемое спин-спиновое взаимодействие между активными ЯМР ядрами. Это взаимодействие возникает в результате переходов между различными спиновыми состояниями ядер в химических молекулах, что приводит к расщеплению сигналов ЯМР. Это расщепление может быть простым и сложным и, как следствие, его либо просто интерпретировать, либо оно может запутать экспериментатора.
Это связывание обеспечивает детальную информацию о связях атомов в молекуле.
[править]Взаимодействие второго порядка (сильное)
Простое спин-спиновое взаимодействие предполагает, что константа взаимодействия мала в сравнении с разницей в химических сдвигах между сигналами. Если разность сдвигов уменьшается (или константа взаимодействия увеличивается), интенсивность мультиплетов образцов искажается, становится более сложной для анализа (особенно если система содержит более 2 спинов). Однако в мощных ЯМР-спектрометрах искажения обычно умеренные и это позволяет легко интерпретировать связанные пики.
Эффекты второго порядка уменьшаются с увеличением разницы частоты между мультиплетами, поэтому высокочастотный ЯМР спектр показывает меньшее искажение чем низкочастотный спектр.